欢迎访问新医改评论 XYGPL.COM 您是第 3628983 位访问者

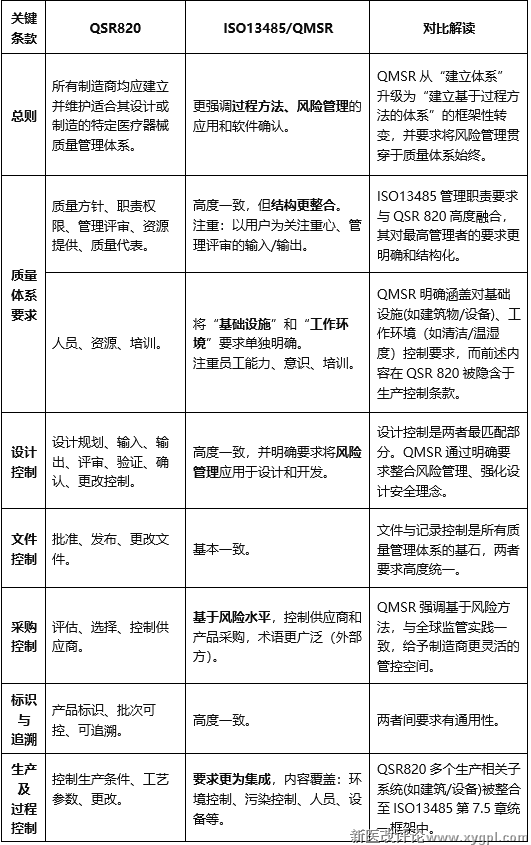
1.总则
QSR820:所有制造商均应建立并维护适合其设计或制造的特定医疗器械质量管理体系。
ISO13485/QMSR:更强调过程方法、风险管理的应用和软件确认。
对比分析:QMSR从"建立体系"升级为"建立基于过程方法的体系"的框架性转变,并要求将风险管理贯穿于质量体系始终。
2.质量体系要求
QSR820:质量方针、职责权限、管理评审、资源提供、质量代表。
ISO13485/QMSR:高度一致,但结构更整合。
注重:以用户为关注重心、管理评审的输入/输出。
对比分析:ISO13485管理职责要求与QSR 820高度融合,其对最高管理者的要求更明确和结构化。
QSR820:人员、资源、培训。
ISO13485/QMSR:将"基础设施"和"工作环境"要求单独明确。注重:员工能力、意识、培训。
对比分析:QMSR明确涵盖对基础设施(如建筑物/设备)、工作环境(如清洁/温湿度)控制要求,而前述内容在QSR 820被隐含于生产控制条款。
3.设计控制
QSR820:设计规划、输入、输出、评审、验证、确认、更改控制。
ISO13485/QMSR:高度一致,并明确要求将风险管理应用于设计和开发。
对比分析:设计控制是两者最匹配部分。QMSR通过明确要求整合风险管理、强化设计安全理念。
4.文件控制
QSR820:批准、发布、更改文件。
ISO13485/QMSR:基本一致。
对比分析:文件与记录控制是所有质量管理体系的基石,两者要求高度统一。
5.采购控制
QSR820:评估、选择、控制供应商。
ISO13485/QMSR:基于风险水平,控制供应商和产品采购,术语更广泛("外部方")。
对比分析:QMSR强调基于风险方法,与全球监管实践一致,给予制造商更灵活管控空间。
6.标识与追溯
QSR820:产品标识、批次可控、可追溯。
ISO13485/QMSR:高度一致。
对比分析:两者间要求有通用性。
7.生产及过程控制
QSR820:控制生产条件、工艺参数、更改。
ISO13485/QMSR:要求更为集成,内容覆盖:环境控制、污染控制、人员、设备等。
对比分析:QSR820多个生产相关子系统(如建筑/设备)被整合至ISO13485第7.5章统一框架中。
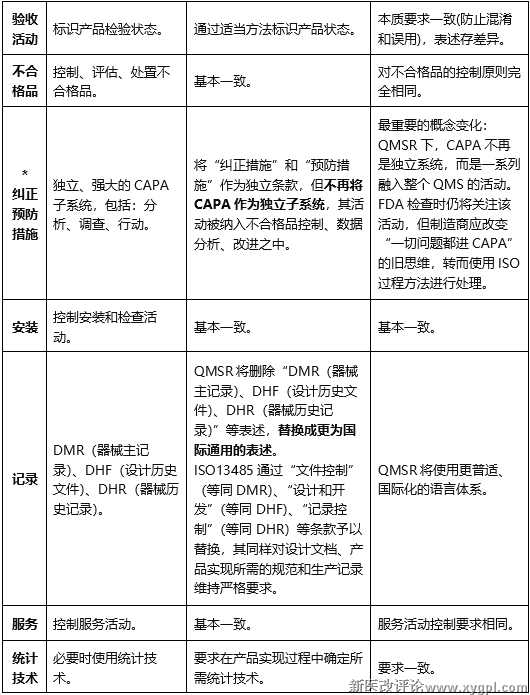
8.验收活动
QSR820:标识产品的检验状态。
ISO13485/QMSR:通过适当的方法标识产品状态。
对比分析:本质要求一致(防止混淆和误用),表述存差异。
9.*纠正预防措施
QSR820:独立、强大的CAPA子系统,包括:分析、调查、行动。
ISO13485/QMSR:将"纠正措施"和"预防措施"作为独立条款,但不再将CAPA作为独立子系统,其活动被纳入不合格品控制、数据分析、改进之中。
对比分析:最重要的概念变化:QMSR下,CAPA不再是独立系统,而是一系列融入整个QMS的活动。FDA检查时仍将关注该活动,但制造商应改变"一切问题都进CAPA"的旧思维,转而使用ISO过程方法进行处理。
10.记录
QSR820:DMR(器械主记录)、DHF(设计历史文件)、DHR(器械历史记录)。
ISO13485/QMSR:QMSR将删除"DMR(器械主记录)、DHF(设计历史文件)、DHR(器械历史记录)"等表述,替换成更为国际通用的表述。
ISO13485通过"文件控制"(等同DMR)、"设计和开发"(等同DHF)、"记录控制"(等同DHR)等条款予以替换,其同样对设计文档、产品实现所需的规范和生产记录维持严格要求。
对比分析:QMSR将使用更普适、国际化的语言体系。
|
|
||||
相关文章

鄂公网安备 42010302000616号
| 新医改评论 版权所有 | 备案/许可证:鄂ICP备10208130号-1 | Copyright @ 2010-2020 xygpl.com All Rights Reserved