欢迎访问新医改评论 XYGPL.COM 您是第 3870593 位访问者

6月3日,美国FDA发布了一份备受关注的指导草案,核心指向如何更高效地将基因编辑等前沿CGT产品带给患者。
草案提出,申办方可以充分利用公开信息与成熟的“平台知识”——涵盖化学、制造与控制(CMC)数据、非临床研究及临床经验——来简化监管申报流程,避免重复验证。生物制品评价与研究中心(CBER)代理主任Karim Mikhail明确表示,此举旨在“加速创新,同时不损害严格科学标准”,让那些患有罕见、危及生命疾病且几乎无药可用的患者能更快获得治疗。
这份文件被行业视为FDA在CGT领域的一次重要“松绑”信号。与之配套的,还有推荐采用新一代测序技术评估脱靶编辑风险的指南草案,以及鼓励企业在产品开发极早期(甚至在新药临床试验IND申请之前)就通过INTERACT会议与监管机构沟通的倡议。
治疗产品办公室代理主任Vijay Kumar进一步解释,利用已有知识并不意味着降低标准,而是要在保持最高安全性和有效性标准的同时,提高整体效率。
然而,如果将视野从华盛顿的监管大楼移向全球药物创新的竞技场,一个更复杂的图景便会浮现。FDA此番提效之举,既是科学监管的进化,也是一场关乎产业竞争力与临床验证速度的博弈中,来自美国一方的主动应变。
而它最强劲的参照系,正在太平洋另一端迅速崛起。
01快车道上的中国
FDA试图用“平台知识复用”来缩短的时间差,在中国已经通过另一种方式被大幅压缩。
根据麦肯锡最新报告,中国已将从早期药物发现到新药临床试验申请的时间线缩短了50%至70%。到2023年,中国在全球临床研究中的份额攀升至39%,在患者招募和开发速度上超越了美国和欧盟。
这种效率优势并非仅靠成本驱动,更深层的原因在于一套独特的临床研究制度安排——研究者发起的临床研究(IIT)。
与必须经过监管机构IND审评的传统路径不同,在中国,由医疗机构研究者自主发起的IIT,仅需通过伦理委员会审批并完成备案(法定时限为5个工作日),即可启动人体研究。这意味着,对于CGT等前沿领域,候选药物进入人体试验的行政等待被压缩至以工作日计的极短窗口,与传统IND审评的数月周期形成鲜明对比。
在过去十年间,中国的IIT数量呈现爆发式增长。截至2024年,相关研究已系统分析了超过1000项IIT。
2026年5月1日,《生物医学新技术临床研究和临床转化应用管理条例》(“818号令”)正式施行。该条例首次以行政法规形式为基因编辑、细胞治疗等前沿技术构建监管框架,在保障规范性的同时,有望进一步大幅缩短前沿技术进入临床的周期。
波士顿咨询公司(BCG)医疗保健业务董事总经理John Wu评价,这种方式让某些创新产品得以“迅速进入人体试验阶段”,绕开了美国式漫长审批的初始门槛。
这条“先临床信号、后正式开发”的链条,将原本线性的长周期流程重构为一个高度并行的闭环,大幅压缩了从靶点发现到价值验证的周期。
某种程度上,正是这种肉眼可见的速度,吸引了跨国药企蜂拥而至。2026年开年仅三个多月,中国本土公司的对外许可交易总额已突破600亿美元,全年极有可能超越2025年1300亿美元的纪录。
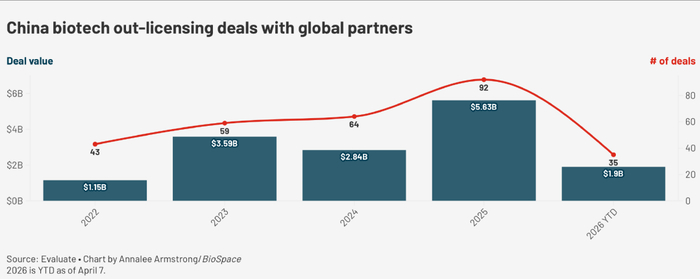
阿斯利康近期宣布投入150亿美元加强在华研发运营,BMS首席研究官Robert Plenge公开承认:“中国一个非常有趣的方面是,我们能够在早期开发中测试更多项目,从而更有效地获得临床概念验证。”
大型药企的需求已经发生结构性转变。Evaluate数据显示,平台型交易正取代单一资产许可,成为中美合作的主流模式。美国公司倾向于许可整个AI药物发现引擎、抗体发现系统或siRNA项目组合,一次协议即获得多次“射门”机会。
相应地,中国生物科技公司凭借早期临床数据争取到更高的首付款(2026年平均首付已达1.67亿美元,同比增长36%),而愿意接受降至个位数的销售分成。这种交易逻辑的本质,是中国以低成本临床验证能力将分子推到价值拐点之上,全球药企则为其支付溢价。
02当科学遭遇审查清单
然而,就在效率逻辑推动中美生物医药深度融合之际,一道政治推力正在反向拉扯。
2026年5月,密歇根州共和党众议员John Moolenaar与民主党众议员Debbie Dingel联合提出《2026年生物技术投资国家安全法案》,要求将生物技术纳入财政部对外投资审查清单,矛头直指中美之间的跨境许可协议、合资企业和股权投资。
Moolenaar点名辉瑞、BMS与中国公司的交易为“危险的交易”,甚至推动国会语言拟禁止在向FDA提交IND申请时纳入来自中国的临床研究数据。
这股保护主义浪潮一旦落地,将从根本上切断中国IIT数据为美国注册试验提供早期证据的路径,让两国临床验证体系走向割裂。
但吊诡的是,FDA自身的监管要求——任何药物获批必须至少有20%的试验在美国境内完成——此前已经制造了一个“美国锚点”:中国可以担任高效的概念验证加速器,但最终的注册证据必须扎在本土。
这种安排曾在效率与监管主权之间取得了微妙平衡,而新法案试图进一步斩断早期数据的流通,极可能将跨国公司的研发逻辑逼入死角。
药企的态度因此变得复杂而分裂。
一方面是科学务实主义——只要采用“同样严格的控制措施”,不论研究在何处进行,数据都可指导全球临床设计。
另一方面则是地缘风险规避,大型CRO公司Charles River Labs的首席执行官James Foster坦承,企业面临重新考虑在华投资的政治压力,但也同样清楚“在中国获批的药物必须在中国进行试验”。
制药巨头们在焦虑中采用“双轨制”:在中国深度嵌入早期研发和临床验证以获取速度优势,同时确保美欧的确证性研究足以满足注册要求。
03让患者定义终局
这一切将全球生物医药引向一个关键问题:临床验证的速度与成本,究竟应该由谁来主导,又受何种规则约束?
FDA在基因编辑疗法领域的监管“松绑”,本质上是用科学认知的进步来换取时间,是在不降低安全标准的前提下做的内部流程优化,但外部竞争的压力并不会因此缓解。
中国制度性赋予临床研究的速度弹性,已经深刻改变了全球药物创新链条的布局。正如Trace Neuroscience公司首席执行官Eric Green所言,中国的崛起显示了临床数据的力量,即使是来自较小患者群体的数据。
与此同时,地缘政治的强制介入,则可能将这种竞争从良性拖入零和。限制美国能够获取的药物与数据,最终代价将由等待新药的患者承担。
欧洲已经感受到类似的阵痛。欧盟临床试验份额从2013年的22%降至2023年的12%,欧盟委员会正通过《生物技术法案》奋起直追,试图将跨国试验授权时间从75天压缩至47天。
全球主要医药创新体都在通过不同的制度工具来争夺“临床验证”这一战略高地,但无论政策如何演变,最不可被辜负的,永远是那些等不及的生命。
Kivu Bioscience公司首席执行官Mo Trikha在最近的行业讨论中形容,面对中国带来的竞争,“我们必须拥抱它,架起一座金门大桥”。这座大桥的两端,连接的不应是脱钩的政治指令,而是基于科学共识的效率竞逐与患者利益。
参考文章:
1、FDA Issues Draft Guidance to Help Accelerate Cell and Gene Therapies for Patients;FDA
2、Deal dynamics between Chinese biotechs and global pharma companies are changing fast, with the biotechs seeking higher upfront payments and the Big Pharmas seeking more expansive platform deals.;biospace
3、No longer a bargain pool, Chinese biotechs command higher premiums;biospace
4、Exclusive: Bipartisan group of lawmakers unveil new bill to restrict China biotech;endpoints
|
|
||||

鄂公网安备 42010302000616号
| 新医改评论 版权所有 | 备案/许可证:鄂ICP备10208130号-1 | Copyright @ 2010-2020 xygpl.com All Rights Reserved